
Scott Hollister, Ph.D.
- Associate Chair of Translational Research
- Patsy and Alan Dorris Chair in Pediatric Technology Professor
- Director, Center for 3D Medical Fabrication
- Director, Tissue Engineering & Mechanics Laboratory
Email
Location: UAW 2102
Phone: 404.385.5506

Adam McCallum, Ph.D.
- Translational Research Advocate
Email
Location: UAW 4109
Phone: 404.385.8531
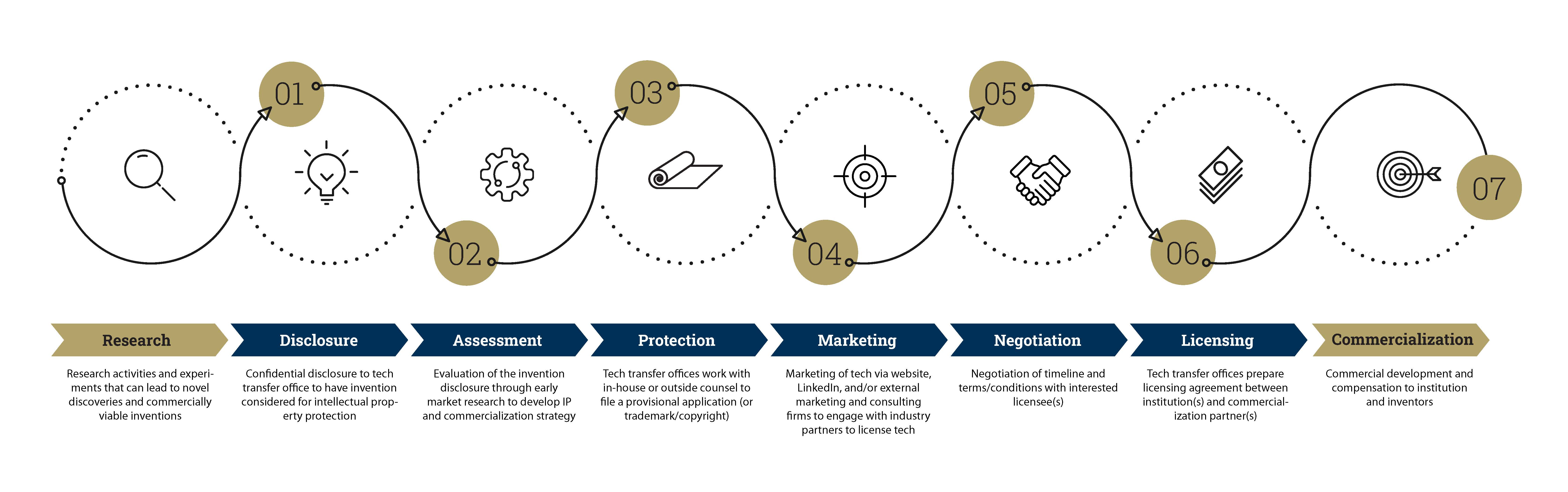
What is a license?
In its most basic terms, a license is simply a permit that is granted by a party to another party as part of an agreement to practice or use something. In academia, when an external party is interested in acquiring the rights to use or develop university-owned intellectual property (IP), the party will license the IP from the university, which entails negotiation and execution of a licensing agreement between both parties. The licensing agreement is a legal contract between the owner of the IP (licensor) and the party who is seeking authorization of the rights (licensee) to use, sell, and/or display the licensor’s IP in exchange for royalties and/or a fee for a specified period of time.
Why is licensing Important?
Licensing IP from academic institutions has become an omnipresent corporate strategy for both the institutions as well as the companies that license the technologies. For academic institutions, licensing can provide a significant source of revenue for the institution. In addition, licensing technologies from institutions can be beneficial from a strategic position for companies who want to add innovative R&D projects to their portfolios and/or minimize R&D costs associated with developing those innovations de novo in-house.
As such, academic institutions are important partners in technology innovation and provide a tremendous impact to communities across the globe that is often enabled by the licensing of academic technologies to external commercialization partners. For academic innovators, this process provides an incentive to develop the technology without the risks and time commitments associated with starting a new venture. Furthermore, companies interested in licensing technologies often have more substantial resources dedicated to increasing the technology readiness level (TRL) enough to progress toward creating a marketable product. However, choosing the right commercialization partner can often be the difference between success and failure.
Choosing the right commercialization partner
The licensing of institution-owned IP from startup companies formed by one or more of the inventors of the IP is commonplace in academic technology transfer. In these cases, the licensing process is relatively straightforward (see the Licensing Intellectual Property to University Startups section below). However, for IP that is being licensed by an external licensor not affiliated with the institution or inventors, choosing the right commercialization partner is crucial for progressing toward the goal of commercializing the technology defined by the IP being licensed. In most cases, when there is interest from a potential licensee, the company will contact the TTO and/or the inventors, and the TTO will vet the licensee and begin negotiations to execute a license agreement with that licensee. However, no matter if there is one potential licensee or multiple at a given time, there are several factors that need to be considered to ensure that the licensor and licensee are compatible enough to ensure (or progress toward) commercialization of the technology.
One factor to consider is whether the licensee has successfully demonstrated the commercialization of (a) related technology(ies) or technologies in the same field, or whether the R&D pipeline of the licensee is complementary to the technology that the licensor is interested in licensing. For example, if you identified an efficacious antiviral therapeutic or vaccine with promising preclinical data, it might be advantageous to license the IP to a pharmaceutical or biotechnology company with expertise in developing antiviral pharmaceuticals.
Another factor to consider is the stage the IP that is being licensed. For technologies that are patented, or in the process of being patented, the scope of already-issued patents and pending applications (including any hurdles that may arise during examination, such as office actions) can affect the terms of an agreement, especially since licensees are expected to pay for patent-related expenses whether proactively or retroactively. Furthermore, a commercialization partner who may only want IP rights in the USA may not want to pay for patent-related expenses if PCT applications were filed in other jurisdictions. The opposite is also true; having US-only rights may be deleterious for licensees who want IP rights in other jurisdictions. While innovators are not directly involved in the negotiation of a license agreement to avoid any conflicts of interest, inventor input is still a valuable factor to consider when the tech transfer office (TTO) selects a licensee, especially since the relationship between parties that involves a technology license lasts for multiple months to several years.
Another extremely important factor to consider is the financial strength of the company. Licensees are typically expected to reimburse the institution for all incurred patent expenses; fund the commercial development of the technology; and pay the licensor royalty fees, milestone payments, and other fees. The type of license agreement that a licensee desires is also an important factor to consider. Depending on the TRL, licensees may request an option agreement, which serves as a guarantee that the institution will not license the IP to another company for the duration of the option period (typically 6–12 months). During that time, the licensee can assess the technology and decide if they would like to license the IP before the option period ends. Exclusivity versus non-exclusivity, as well as which fields of use are granted to the licensee, must also be considered during the licensing negotiations.
Ideally, innovators should be devising a commercialization strategy before filing an invention disclosure with their TTO. That commercialization strategy can be further refined with the TTO during patent prosecution, while the technology is being further developed, and/or based on feedback while marketing the technology to potential licensees. Importantly, it is highly advantageous to define an endpoint and work backward to develop a commercialization strategy. That way, choosing a licensee that best aligns with that strategy or who has the resources and vision to develop a commercialization plan that benefits all parties will facilitate the commercialization process.
Licensing agreements
The TTO at your primary institution—who, in this case, is either Georgia Tech or Emory—is responsible, among many tasks, for negotiating and executing licensing agreements between the institution and licensees. The vast majority of license agreements are unique and will have different terms; while institutions often have license agreement templates, the terms of each agreement are always negotiated between parties to carve out an agreement that is specific for that case.
These terms are important because any unclear clauses in the agreement can result in future litigation between the parties involved in the agreement. However, the terms of the licensing agreement may be shared with the innovators once the license has been executed. For more information on what terms are typically defined in a licensing agreement, see the Terms of a License Agreement section.
As mentioned under the Basics of Technology Licensing section, a license agreement is defined as a contract between two parties that gives permission to a party (licensee) to use, sell, and/or display the licensor’s IP. Generally, the five main types of license agreements are:
- Patent license
- Software license
- Trademark license
- Copyright license
- Trade secret license
The vast majority of license agreements that are executed between academic institutions and external companies in the biotechnology field are patent licenses, which gives certain rights to the intellectual property (IP) defined within one or more patents to the licensee for a determined period of time. Those rights are negotiated between the licensee and licensor and are unique to the individual license agreement, depending on the interests of the external company (and, in many cases, the interests of the innovators). Below is a list of the most common types of licenses, with an emphasis on patent licenses, executed between Georgia Tech/Emory and licensors.
- Exclusive license: grants exclusive rights of use to one or more parties to use the IP defined within one or more patents being licensed. Under an exclusive license agreement, the licensor agrees not to grant to any other licensees any of the same rights that fall within the scope (or field(s)) of the license agreement.
- Non-exclusive license: grants to the licensee the rights to the IP defined within one or more patents being licensed but also allows the licensor to grant licenses to other parties.
- Option license: grants the opportunity to an industry partner to evaluate a technology for a given period of time before making the decision to license the technology. Essentially, an option is a promise that the university will not license the technology in the defined field of use to another company during the term of the option agreement. However, an option does not give the company the right to commercially practice the IP. While options are risk-averse, they are limited to the term of the agreement.
The greatest [initial] hurdle to technology licensing is arguably identifying a potential licensee. However, there are a number of different mechanisms by which innovators can identify and connect with potential licensees, whether it be through active or passive interactions between licensors and potential licensees. Below are several examples of opportunities that academic innovators have to identify and connect with potential licensees.
- Conferences, trade shows, and scientific meetings: Conferences, trade shows, and scientific meetings are often the most important and successful means of showcasing innovative technologies to generate interest from potential licensees. These meetings are attended by stakeholders from academic institutions, non-profit research organizations, (bio)technology companies, healthcare organizations, patent law firms, and myriad other entities, providing ample opportunities for stakeholders to learn about new, innovative research and prototype development coming out of academic institutions.
- Industry-sponsored research: Industry-sponsored research can be an excellent mechanism to increase the chances of external companies licensing university-owned IP. With respect to the innovation life cycle, industry-sponsored research (i.e. applied research) is a step beyond “basic research” that is typically government-sponsored (i.e. through government grants). Therefore, the motivation behind industry-sponsored research agreements is to translate basic science research into a marketable product that benefits society. The sponsored research agreement (SRA) is most often initiated by the university on its terms, and any IP that is generated from the SRA is typically jointly owned. However, the industry sponsor generally has the rights to license the portion of the IP that is owned by the university.
- Technology Scouting: Tech scouting is a process that involves identifying and evaluating emerging technologies to increase partnership and/or licensing and acquisition opportunities for technologies that may have commercialization potential. This method is employed by both academic institutions and industry to drive strategic planning, promote partnerships and collaborations, expand R&D portfolios, and ultimately reduce R&D costs. Academic institutions often employ tech scouting internally to evaluate the commercialization potential of developing technologies in collaboration with the institution’s TTO. Industry also utilizes tech scouting to evaluate academic research to initiate collaborations and license IP from academic institutions.
- Active Marketing: While the first three types of mechanisms to discover and connect with potential licensees are all types of active marketing, this particular example is a variation of cold-calling or emailing potential licensees to inform them of available technologies that they may be interested in. This strategy is often employed by both the technology transfer office (TTO) and the innovators; however, upon disclosure of the invention to the TTO, be sure to make the TTO aware of competitors and potential licensees for the particular technology to facilitate the marketing process. In addition, both Georgia Tech and Emory utilize external tech transfer management firms as resources to increase the exposure of available technologies to potential licensees through a number of different avenues, including advertising the technologies on social media, the creation and management of websites, and networking. These firms include TreMonti and Fuentek.
- Passive Marketing: In the context of technology transfer, passive marketing entails the advertisement of technologies available for licensing to the public through various avenues, including databases, social media, and other online digital solutions that serve to connect academic innovators with industry stakeholders. Below is a list of several passive marketing services that are used by Georgia Tech and Emory:
- Emory OTT and Georgia Tech OTL websites: Both tech transfer offices feature available technologies on their websites.
- LinkedIn: Emory and Georgia Tech both utilize LinkedIn for advertising technologies that are available for licensing.
- IN-PART: IN-PART is an online digital marketing platform that aims to connect academic innovators with industry stakeholders. Licensing associates will ask innovators at various stages of patent prosecution if they would like to have their technologies marketed on IN-PART.
Technology marketing is integral to the licensing process. Marketing exposes potential licensees to the technologies that are currently available for licensing. Because licensing is not limited to a particular stage of the patent and copyright prosecution process—meaning, a technology can be licensed at any stage of prosecution and examination, including immediately after an invention disclosure (ID) is filed with the primary institution’s technology transfer office (TTO)—marketing can, in theory, be conducted any time after the TTO receives an ID.
However, marketing activities at Georgia Tech and Emory are typically initiated 6–9 months into the provisional patent stage, when the licensing associates request a data review with the innovators to assess strategy for future activities, including converting the provisional application to a non-provisional application. It is important to note that marketing is voluntary; some innovators may choose to keep their technologies undisclosed to the public, at least until non-provisional applications are published. Therefore, marketing is left to the discretion of the innovators.
At Georgia Tech, a number of mechanisms are used by the Office of Technology Licensing (OTL) to market currently available technologies. The licensing associates at Georgia Tech OTL manage the content posted to the online subscription platforms and liaise with external marketing and IP management firms. Marketing mechanisms utilized by Georgia Tech OTL include:
- LinkedIn: LinkedIn has become a ubiquitous mechanism by many research institutions to advertise their technologies currently available for licensing.
- Fuentek, LLC: Fuentek is an external technology transfer consulting firm that Georgia Tech uses for several different technology transfer services, including creating marketing materials for technologies in the OTL system, assessing the commercialization potential of technologies, and building relationships between researchers and stakeholders.
- TreMonti: TreMonti is another external innovation management consulting firm that Georgia Tech utilizes for marketing and commercial engagement activities, including the generation of non-confidential marketing summaries, active marketing services, and networking and training opportunities.
- IN-PART: Georgia Tech utilizes IN-PART as an intelligent online matchmaking platform for connecting companies and other organizations with Georgia Tech innovators and their technologies. Georgia Tech OTL directly uploads disclosures to the closed-loop portal that is only accessible to industry decision-makers who subscribe to the service so that these stakeholders can have direct access to these disclosures for making business decisions.
- Active marketing: Active marketing is utilized by both licensing associates and innovators. This method of marketing includes reaching directly out to potential licensees to inform them of technologies that may be relevant to their R&D pipeline or that they may have interest in licensing.
The Emory Office of Technology Transfer (OTT) utilizes most of the same marketing resources as Georgia Tech, except for Fuentek, LLC. However, Emory OTT has in-house marketing associates who are responsible for managing the content uploaded to LinkedIn and IN-PART, creating marketing summaries, and other related marketing activities.
As stated in previous sections, licensing agreements are unique to each case, and typically no two licensing agreements will look the same. However, both Emory and Georgia Tech have model licensing agreements that they use as a basis for negotiating licenses with licensees. Given that license agreements are legal contracts, the licensee will be required to abide by terms and meet certain milestones that are negotiated by both parties and that are defined in the agreement for the term of a license, otherwise the license can be terminated by the licensor. Key terms that comprise an IP license agreement include (but are not limited to):
- Recitals: identifies the parties involved in the agreement, the purpose of the agreement, the IP that is being licensed from the licensor by the licensee, and the scope of the rights to the IP being licensed.
- Type of License Agreement: Exclusive, non-exclusive, option, etc.
- Term and Termination: defines the length of the license, what are the grounds for termination of the license by each party, and what remedies each party are entitled to upon breach of the contract.
- Financials: what payments in the form of royalties, milestone payments, maintenance payments, patent reimbursement costs, etc. must the licensee pay the licensor and how frequently throughout the term of the license.
- Fields of Use: what fields of use are the licensees entitled to maintain the rights to use the IP? For example, if the owner of the IP is licensing the rights to commercialize a therapeutic for breast cancer to a licensee, the fields of use may include (or be limited to) breast cancer and precancerous lesions of the breast tissue.
- Milestones: milestones establish objectives on which both parties agree that are designed to guide the development or commercialization of the technology being licensed. Upon completion of each milestone, the licensee is typically required to pay the licensor a milestone payment that is defined in the agreement.
Commercializing technologies developed as a product of academic research is a daunting task, whether the end goal is to license the technologies to an external (bio)technology company or to start a (bio)tech venture based on the technology. However, for those who choose to venture down the path toward becoming an entrepreneur, several considerations should be made to increase the chances of success. BME researchers who are looking to start a company should discuss their strategy and goals behind starting the venture with the Translational Research Advocate, a resource in BME who can help guide them with important considerations for a startup and identify the resources that are most applicable to their startup. Several of these considerations are discussed below.
Planning for a Startup
Why a Startup?
New venture creation is a challenging and time-consuming endeavor even for an experienced entrepreneur. Therefore, an integral question to ask yourself is, “why a startup?” In other words, what are the goals that the innovators hope to accomplish with the startup and would it be easier to accomplish these goals by licensing the technology to another company? Being an entrepreneur requires a different mindset than being a scientist, and it is important to ask yourself whether you truly want to venture into the world of being an entrepreneur.
Intellectual Property (IP) and Compliance
Before starting a (bio)tech venture, it is important to have a conversation with your Technology Transfer Office (TTO) about which invention(s) you are wanting to commercialize. Because TTOs manage university-owned inventions (i.e. IP), meeting with the TTO will enable you to understand the institution’s policies on startup formation and licensing the IP from the institution. They can also handle situations surrounding jointly owned IP with other institutions and/or research sponsors. It is also important to contact the Office of the General Counsel and Office of Research Integrity Assurance (oria.gatech.edu) at Georgia Tech and/or the Conflict of Interest and Commitment Office (Emory) to declare any potential conflicts of interest and submit/update any activities, as institutions require that employees notify their compliance offices when they undertake external commitments that may interfere with the primary obligations of the employees. These entities will also help set up appropriate agreements if the startup will need access to University resources.
Startup Consultations
Emory and Georgia Tech have several organizations, programs, and resources available to their faculty, staff, and students to launch and support startups. At Georgia Tech, after meeting with the Office of Technology Licensing (OTL) and Office of the General Counsel about intentions to start a company, it is recommended that you speak to Cynthia Sundell and Harold Solomon, two Principals at VentureLab who have decades of experience in industry and startups in the life science, medical device, and software sectors. VentureLab comprises a team of experts in entrepreneurship who collaborate with faculty and staff to create startups from university-owned IP.
At Emory, Faculty & Startup Services (FSS), which is a program that was established by the Office of Technology Transfer (OTT) to provide the Emory community with resources and assistance for launching and supporting startups, is a valuable resource for Emory faculty, staff, and students and provides startup services similar to Georgia Tech’s VentureLab. The current director of FSS is Patrick Reynolds. Patrick, as well as Harold and Cynthia, can guide researchers through every aspect of new venture creation, from ideation all the way through company formation and recruiting a C-suite. Their contact information, as well as links to VentureLab’s and FSS’s websites, are provided below.
After consulting with entrepreneurship and IP experts, if starting a company is truly the best fit for your technology(ies), and the skillset of the team is diverse enough to develop the technology and acquire funding to support the development and growth of the business, the next step is to dive into customer discovery to identify whether the problem that you are trying to fix really exists and what are the applications for the technology(ies).
Check out VentureLab and Faculty & Startup Services for more information on what services they provide.
Set up consultations with VentureLab or Faculty & Startup Services:
Cynthia Sundell (Principal at VentureLab): Cynthia.sundell@ibb.gatech.edu
Harold Solomon (Principal at VentureLab): Harold.solomon@venturelab.gatech.edu
Patrick Reynolds (Director, Faculty & Startup Services): Patrick.reynolds@emory.edu
Customer Discovery
Customer discovery is a fundamental (and iterative) process when starting a company because it enables entrepreneurs to assess whether the technology has a real value to society. In addition to poor management, startups often fail because there is no market fit for the product. Therefore, surveying whether customers have a need for the product that the startup is commercializing is crucial for designing a business strategy. There are a number of different mechanisms for customer discovery.
For academic innovators, both the NIH and NSF have Innovation Corps (I-CorpsTM) programs, which are immersive training programs for entrepreneurs to learn about the customer discovery process from various experts in the biotech sector and conduct preliminary market research on their technologies. To be eligible for the NIH I-CorpsTM program, a company must be incorporated, and that company must have received an SBIR/STTR grant through one of the institutes at the NIH. For NSF I-CorpsTM eligibility, a team must have some ties to the NSF, whether it be through receiving a grant from the NSF (within the past five years) or participation in an I-CorpsTM Sites program (more information is provided under the Market Research and Customer Discovery section).
Georgia Tech is one of the four institutions that make up the NSF I-CorpsTM South Node and is a hub for the NSF I-CorpsTM Sites program, which is a regional arm of the national NSF I-CorpsTM Teams program. These programs also help entrepreneurs focus their business plan to facilitate the translation of an invention to a marketable product. Furthermore, Emory OTT, in partnership with the Georgia Clinical and Translational Science Alliance (CTSA), created the Kauffman FastTrac® TechVentureTM entrepreneurship training course that is designed to teach researchers how to grow an idea into a successful business. There are numerous other resources available at both Georgia Tech and Emory to help faculty, staff, and students with new venture creation. These resources are summarized in the Commercialization Ecosystems section.
Forming a Startup
Choosing a business structure
The first step in launching a startup entails incorporating the company and choosing which legal entity the company will use (i.e., C-corps, S-corps, LLC, etc.). While this can be done online yourself, it is recommended to hire a lawyer or certified accountant to understand the differences between the structure of each entity, what tax implications each entities has, and how to set up the initial equity structure of the company. Most companies incorporate in Delaware, for example, because companies that are incorporated in Delaware but do not do business in Delaware do not have to pay corporate income taxes. Note: Because there are both legal and tax implications involved in starting a company, universities cannot provide legal advice on what the best business strategy is for each venture. However, VentureLab, FSS, the TTOs, and other commercialization-focused entities at both institutions can provide entrepreneurs with a number of different external resources that have expertise in these areas and with which university startups have had success in the past. The Internal Revenue Service (IRS) has also created a Checklist for Starting a Business to guide entrepreneurs through the basic steps of starting a business.
Building a team
After the company has been incorporated, the next-most-important step is to form the team. Choosing the appropriate management of the company can be the difference between success and failure; as such, investors such as venture capitalists and angel investors more often choose to invest in a strong team of people than in the technology (to some extent). Choosing which people are going to run the company is often the most difficult part of starting a company, as there needs to be a delicate balance between scientific expertise and business acumen. In the very early stages of launching a startup, especially in the (bio)tech sectors, the scientists and/or technologists who founded the company are often the ones running the company. For example, principal investigators will often have graduate students and/or post-docs who developed the technology that led to the formation of the company guide the scientific and technological development for the company as CTOs, CSOs, etc. In addition, they may even be the face of the company until a person with business expertise has been identified to lead the company.
Until then, it is often recommended to utilize business consultants or advisors who can help make business decisions for the company and identify a suitable management team. Both VentureLab and FSS, as well as other organizations, such as ATDC, are valuable resources that can aid in identifying executive-level managers (C suite). If any of the personnel of the startup are also university employees, the personnel—whether (under)graduate students, post-docs, research staff, or professors—need to discuss their time commitments and responsibilities with each institution’s compliance office to ensure that any individual or institution COIs are reported and managed properly.
Acquiring funding
Financing a startup is pivotal for ensuring that the startup can operate effectively. In the early stages of a startup, acquiring non-dilutive funding will ensure that your company receives capital to pay employees and invest in R&D without relinquishing any equity or ownership in the company. There are various types of non-dilutive funding that startups can acquire, but grants are by far the most common for academic startups. A number of different commercialization grants are available to Emory and Georgia Tech faculty, staff, and students pre- and post-startup formation.
Pre-startup formation, the Georgia Research Alliance (GRA) and Biolocity grants, as well as NSF I-CorpsTM program funding, are among the most popular seed grants for members of the BME community. Post-startup formation, SBIR/STTR grants and the NIH I-CorpsTM program provide additional non-dilutive funding opportunities to incorporated companies. The Advanced Technology Development Center (ATDC), Georgia’s technology business incubator whose headquarters are located on Georgia Tech’s campus, can provide free SBIR/STTR grant consulting to its members. In addition, GRA and SBIR/STTR grants have multiple phases to allow researchers and/or companies to receive multiple rounds of funding upon successful completion of previous phases. See the Early-Stage Commercialiization Funding section for more details, contact information, and links to apply.
Additional considerations
Before starting a company, try to keep the development of the technology in the lab for as long as possible. This is because, once a company is formed, further development of the technology for the startup company becomes a conflict of interest (COI). Establishing a clear demarcation between work/personnel for academic research purposes and for your company is imperative for mitigating potential COIs. In addition, balancing personal life and work life can be challenging, especially for professors, who are required to teach, manage a research lab, and tend to other administrative responsibilities that are associated with academia. Therefore, taking a reduction in workload or a leave of absence to focus on growing a startup company may be necessary. It is important that you assess what level of personal risk you are willing to take before starting a company.
Often called “the valley of death” in technology transfer, the journey from taking an innovative idea to a prototype or proof-of-concept requires significant funding, and acquiring enough funding is often a tremendous obstacle that hinders (bio)technology innovation. Whether a company has been formed or not, there is a number of different funding mechanisms available that can provide both non-dilutive and dilutive funding to early-stage commercialization teams to bridge this gap. This section serves to highlight several seed grants and federal grants applicable to members of the Biomedical Engineering community at Georgia Tech and Emory to fund the development of proofs-of-concept and prototypes.
Seed Grants
Georgia Research Alliance (GRA): GRA is a state- and private-funded nonprofit organization whose mission is to grow Georgia’s economy by strategically investing in innovative research to promote the translation of technologies that benefit communities across the world and enrich the economy and reputation of Georgia. The commercialization arm of GRA provides funding that is divided amongst four phases to seed Georgia-based startups.
Phase 1: Up to $50,000
- Applications are received on a rolling basis and evaluated once per month. Funds are distributed through your lab account and done in two traunches (Phases 1a and 1b).
Phase 2: Typically ~$100,000, but is flexible based on need and matching grant value
- This phase requires a matching grant. Biolocity (see below) often serves as the matching grant for BME-related technologies. Applications are received on a rolling basis.
*Phases 1 and 2 do not require the formation of a company, and funds are distributed to the awardees through their university.
Phase 3: Up to $250,000 in the form of a loan (not a grant)
- This phase of funding is only available to startup companies based in Georgia who have completed the first two stages of GRA funding.
Reach out to Cindi Sundell, cynthia.sundell@venturelab.gatech.edu, who will help you through the application process. Further information on the different phases of GRA funding and application templates can be found on Georgia Tech VentureLab’s website.
Biolocity: Biolocity provides a combination of funding, project management, and consulting resources to early-stage technologies that address an unmet clinical need and have compelling commercial potential. This includes innovations within any clinical discipline with the ability to impact patient health. Funds are provided as justified, but typically range from $100,000–150,000, and typically 4–6 projects are funded annually. The application timeline is typically six months and involves the following steps:
- RFP announced; Biolocity will meet individually with each project submitted (November–December); timeline varies slightly year-to-year.
- Invitations for full applications sent out (January)
- Down-selection and invitation for five-minute mini pitch applications sent out (Mini pitches performed in March in front of Biolocity Oversight Committee)
- Down-selection and invitation for full applications sent out (March-April)
- Full ten-minute pitch performed before Biolocity Oversight Committee (May-June)
- Decisions made on funding (June–July)
*Biolocity funding can serve as a matching grant for the Phase 2 GRA grant.
NSF I-CorpsTM: The goal of the NSF Innovation Corps (NSF I-CorpsTM) program, created in 2011 by the NSF, is to reduce the time and risk associated with translating promising ideas and technologies from the laboratory to the marketplace. The NSF I-CorpsTM program uses experiential learning of customer and industry discovery, coupled with first-hand investigation of industrial processes, to quickly assess the translational potential of inventions. The program entails a six-week intensive customer discovery program and provides $50,000. This program is very useful for faculty thinking of forming a startup company around the intellectual property being developed in the lab; company formation is not a prerequisite to participation in the program. Visit the NSF I-CorpsTM website for more information on how to apply and eligibility requirements.
Health Systems Institute (HSI) Research Seed Grant: HSI is a collaboration between CHOA, Georgia Tech College of Engineering, and the Georgia Tech College of Computing that provides seed grant funding to support the development of collaborative and interdisciplinary research projects with commercial potential. Each award is up to $50,000 per year, and proposals are typically due in June, with funding to begin in August. For eligibility, teams must consist of a faculty member from Georgia Tech or Emory or a staff member from CHOA.
Emory Office of Technology Transfer (OTT) Proof-of-Concept Fund: The OTT POC fund provides Emory researchers and clinicians with small seed grants that range from $5,000–$30,000 to accelerate medical technology commercialization. The fund is managed by Emory OTT, and eligibility for the seed grants requires that the Emory inventions be unlicensed and disclosed to OTT. Approximately 3–5 investments are made per year.
Federal Grants
Small-Business Innovation Research (SBIR) & Small Business Technology Transfer (STTR) Programs: SBIR and STTR programs are highly competitive programs that encourage domestic small businesses to engage in Federal Research/Research and Development (R/R&D) with the potential for commercialization. SBIR/STTR applications are typically due in January, May and September. A number of different federal agencies provide SBIR funding—including the NIH, NSF, CDC, FDA, and DOD, to name a few—in three phases:
- Phase I, whose objective is to establish technical merit, feasibility, and commercial potential prior to seeking Phase II funding, provides approximately $50,000–$250,000 for 6 months (1 year for STTR programs).
- Phase II is aimed at providing funding to continue R/R&D efforts that were initiated in Phase I, and funding is based not only on the results that were achieved during the first phase but also the commercial potential and technical merit outlined in the Phase II application.vPhase II awards are typically ~$750,000 for 2 years.
- Phase III does not provide additional funding but is intended to further the commercialization objectives established in Phases I/II.
The Small Business Association’s (SBA) SBIR/STTR website provides a wealth of additional information available on SBIR/STTR programs, including eligibility, funding amounts, the agencies participating in the programs, and beyond https://www.sbir.gov/about. In addition, the Advanced Technology Development Center (ATDC) provides valuable SBIR/STTR consulting services to Georgia entrepreneurs who are interested in applying for funding. Reach out to Connie Casteel at connie@atdc.org to schedule a meeting to discuss these programs. At Emory, the Georgia Clinical and Translational Science Alliance (Georgia CTSA) manages the BizGrants service, which provides academic investigators with advice on how to submit effective SBIR/STTR applications. To schedule a consultation, visit the BizGrants website.
National Science Foundation Partnerships for Innovation (NSF PFI) Grants: The NSF PFI program provides funding to innovators at academic, nonprofit, and public organizations to accelerate the development of breakthrough technologies. The program comprises two broad tracks: the Technology Translation (PFI-TT) track and the Research Partnerships (PFI-RP) track, each of which has specific goals. PFI-TT awards provide up to $550,000 for 18–24 months (15–35 awards), whereas PFI-RP awards provide up to $1,000,000 for 36 months (10–20 awards). More information on the program, including eligibility, contact information, and resources, can be found at the NSF PFI website.
Dilutive Funding – Angel and Venture Capital Investments
The aforementioned programs offer non-dilutive commercialization funding in the form of grants (and sometimes loans), enabling researchers and innovators to develop their technologies without giving up ownership of their company (if one has been formed). However, there is only so much non-dilutive commercialization funding available to academic researchers and the startup companies that they form. At some point, entrepreneurs will need to pursue angel and venture capital funding as the startup’s seed capital dwindles. These are both dilutive funding mechanisms, which requires giving up partial ownership of your company in exchange for a financial investment in the company. In the Atlanta area, the two most notable angel investment organizations are the Seraph Group and the Atlanta-Technology Angels. There are numerous venture capital firms that have close ties with Georgia Tech and Emory that fund a broad spectrum of innovations. For more information on these, contact your technology transfer office if you are interested in pursuing angel and/or venture capital funding.
After successfully incorporating a company and acquiring funding—seed grants, federal grants, and/or investor capital—to begin commercialization of a technology, the startup must license the intellectual property (IP) surrounding the technology from the academic institution(s) in order to be able to commercialize the technology. This section reinforces the concepts of IP ownership and discusses what startup licenses are and how they differ from license agreements that are executed between mature, established companies and academic institutions.
Who owns the IP?
As employees of either Georgia Tech or Emory and conducting research that leads to the creation of an invention in university-owned facilities, Georgia Tech or Emory owns the IP that is generated as a product of research and research-related activities and is responsible for managing the IP. While mature, established companies are valuable licensing partners for academic institutions, Georgia Tech/Emory startups are equally—if not more—important partners, and the technology transfer offices and other commercialization-focused organizations and programs encourage and support innovators to develop and commercialize early-stage technologies through the formation of startups.
Startup licenses
To encourage researchers to launch startups and to support the startups in their commercialization endeavors, Georgia Tech and Emory offer startup licenses that are intended to streamline the licensing process and reduce the upfront financial burden from patent-related expenses and other obligatory payments to the institution that owns the IP over the term of the license.
Differences between licensing technologies to university startups versus a mature company
Startup licenses are designed to simplify and expedite new venture creation and development. These licenses are often standardized and non-negotiable, but the terms are very favorable, and the license is streamlined for quick execution to incentive innovators at Georgia Tech and Emory to launch starts from university research. Standard startup licenses are typically exclusive licenses, but nonexclusive startup licenses can be drafted if the licensee and licensor agree that is the best option for the particular IP that is being licensed. Option licenses are not common for university startups because the founders who started the company are most often licensing the IP that they developed in their academic laboratories. Comparably, license agreements between established companies and academic institutions can be exclusive, non-exclusive, or exclusive/non-exclusive options, depending on the needs of the company, the maturity of the technology (i.e. the technology readiness level), and other factors. In addition, these license agreements require the licensee to pay previous and on-going patent expenses related to the IP they are licensing, license issue fees, and milestone payments, some of which are often either deferred or waived for startup licenses.
Are you interested in a startup license for your venture?
For those who are new to entrepreneurship, it is recommended that inventors meet with the BME translational research advocate, as well as VentureLab (Georgia Tech) or Faculty & Startup Services (Emory), while engaging with their technology transfer office (TTO) to consult them about what resources are available both at the institutional level—as well as in the local, state, and federal levels—for new venture creation, especially for commercialization funding and customer discovery. While the TTO will engage with the founders of startups often before the venture is created, startup licenses are not typically executed until the venture has a management team other than the principal investigator that can represent the company to avoid any conflicts of interest. Licensing associates in the TTO will reach out to a member of the C suite of the company to request a commercialization plan and get details about the company, including history of the company, financial status and obligations, and what the objectives of the company are how the IP will facilitate those objectives, after which the license associate will facilitate the execution of the license with the startup.
Determining whether the technology you are developing has a true value to society and whether there is a market fit for the technology are critical processes in which startup founders must invest very early on. Customer discovery allows us to identify the appropriate customer segment(s) for a technology product and align the product with the needs of its customers. The vast majority of startup technology companies fail because they invest thousands of dollars into product development when there is no market need for their products. Since new technologies are inherently fluid in their design and their purpose, investing in customer discovery for a product early on in its development and continuing the customer discovery process through multiple iterations will ultimately guide the development of the technology and ensure greater product-market fit. However, customer discovery is not necessarily restricted to new product development; it is also frequently used by companies for assessing whether to target new personas for a particular product or whether to enter a new market.
What is the relationship between market research and customer discovery?
Fundamentally, market research entails identifying trends in target markets and gathering demographic information about customers to understand potential opportunities and limitations. These data are extremely valuable when assessing not only which markets to target but also how large the markets are and who are the target customers. Remember, it is not necessarily about identifying how painful the problem is—seemingly enough, the technology you are developing likely addresses that—but rather whether the problem is large-enough to make it worthwhile to develop a product to solve that problem. Therefore, market research tends to look at the big picture, whereas customer discovery is much more focused and specific because it entails understanding the pain points that customers are currently experiencing with existing solutions (or lack thereof) and identifying opportunities to address those pain points.
Market research: insights and resources
There are numerous companies that conduct marketability assessments, some of which Georgia Tech and Emory utilize for preliminarily determining the commercializability of university-owned IP. In addition, there are a number of market research companies from which you can purchase market research reports online. However, these market reports tend to be very expensive and provide outdated and/or generalized data. In addition to publicly available databases and search engines, Georgia Tech and Emory subscribe to several databases, from which preliminary market research data can be accessed, that can enable you to get a general idea about market trends, barriers to market penetration, and market size. Details about these databases are available in the Market Research document under the Step 1 of the Commercialization Process section.
Customer discovery: insights and resources
Unlike market research, which is heavily data-driven, customer discovery is far more experiential. That is not to say that customer discovery is not data-driven, but it is rooted in understanding the customers’ pain points and identifying a solution to solve those pain points. This needs to be done at the source: with actual potential customers. Therefore, defining and ranking target personas (i.e. specific groups of representative customers) should be the first step in the customer discovery process. Market research (see section above) can be a valuable mechanism for identifying target personas. Once you have identified target personas, the next critical step is to do direct customer discovery research. This can be done through a number of different mechanisms:
- In-depth interviews: use interviews with potential customers to validate any assumptions about unmet needs that customers may have with existing products/services. These interviews should be used to discover and learn about pain points by asking open-ended questions and listening to the needs of the customer rather than pitching/selling your idea and using biased questions.
- Surveys/questionnaires: surveys and questionnaires can often achieve similar results to in-depth interviews but at a more superficial level. Questions on customer discovery surveys should be focused on identifying customer demographics and lifestyles as well as their pains with currently available options. However, be mindful that this mechanism is limited in its scope and may not provide as detailed of information as interviews.
- Focus groups: focus groups entail moderated conversations with a small group of target users that are aimed at identifying their pain points with existing products, understanding the customers’ point-of-view, learn information on their consumer habits, and identifying potential solutions based on their feedback.
At Georgia Tech and Emory, as well as in the Metro Atlanta area, there are a number of government- and institution-sponsored programs that teach nascent entrepreneurs how to do customer discovery and even provide funding to facilitate the process. Listed below are several of those programs, but be mindful that this list is not entirely comprehensive.
- NSF I-CorpsTM Sites Program @ Georgia Tech: The I-CorpsTM Sites program is open to Georgia Tech students, faculty, and staff who are aiming to commercialize their research. The program, which is run by Georgia Tech VentureLab, teaches early-stage commercialization teams how to conduct early customer discovery research and provides $3000 in travel reimbursement for their customer discovery research. This program also serves as “NSF lineage” for the national NSF I-CorpsTM program, more information on which is provided below. Reach out to Melissa Heffner at melissa.heffner@venturelab.gatch.edu for more information on the program. Click here to apply for the Sites program (hyperlink: https://venturelab.gatech.edu/node/16)
- National NSF I-CorpsTM Program: The mission of the national NSF I-CorpsTM program is to improve the quality of startups created from NSF-funded research and teach nascent commercialization teams about entrepreneurship, with emphasis on building business models and conducting customer discovery research. The program entails a 6-week customer discovery bootcamp and includes a $50,000 grant from the NSF to facilitate grassroots customer discovery. Click here for additional information about the national NSF I-CorpsTM program (hyperlink: https://new.nsf.gov/funding/initiatives/i-corps/about-i-corps)
- Eligibility: NSF lineage (recipient of NSF grant in the past five years and/or participation in the NSF I-CorpsTM Sites program) and a team that consists of a technical lead, entrepreneurial lead, and a volunteer industry mentor. Learn more about the eligibility requirements for the program (hyperlink: https://new.nsf.gov/funding/initiatives/i-corps) and get answers to frequently asked questions (hyperlink: https://www.nsf.gov/pubs/2017/nsf17083/nsf17083.jsp)
- NIH I-CorpsTM Program: the NIH I-Corps program is an entrepreneurship training program for companies that have received a small business innovation research (SBIR) or small business technology transfer (STTR) grant from either the NIH or the CDC. This program is an intensive, 8-week bootcamp that is designed to help companies focus their business plan and conduct customer discovery research, and it includes a $55,000 grant. Click here https://seed.nih.gov/I-Corps-at-NIH for more information about the NIH I-Corps program and how to apply.
- CREATE-X Startup Launch: Startup Launch is a summer accelerator program created and operated by CREATE-X that helps students, faculty, and staff transform an innovative idea or prototype into a startup. Startup Launch is a 12-week summer program, and successful teams receive a $20,000 investment to assist with customer discovery, patent protection, and other startup-related activities. Students can also take a for-credit course called Startup Lab, which is geared to help students learn about entrepreneurship and build functional prototypes of their innovative ideas. Click here to learn more information about Startup Launch.
FDA Regulatory Strategies and Approval
The Food and Drug Administration (FDA) is the governing body responsible for regulating, as well as monitoring the safety of, medical devices and drugs in the United States. However, before medical devices and therapeutics can legally be sold in the US, they must be approved (or cleared) by the FDA, before which sufficient evidence must be provided demonstrating that the device or therapeutic is both safe and effective for its intended use.
FDA Regulation of Medical Devices
As stated in Section 201(h) the Federal Food, Drug, and Cosmetic (FD&C) Act of 1938, a medical device is “any instrument, machine, contrivance, implant, in vitro reagent that is intended to treat, cure, prevent, mitigate, diagnose disease in man.” Medical devices are intended to affect the structure or function of the body of humans and animals but do not achieve their primary intended purposes through a chemical action. In addition, their primary intended purposes are not achieved upon metabolism of the device. Medical devices can be as simple as a tongue depressor or as complex as an artificial heart. In 1976, Congress amended the FD&C Act to include regulations for medical devices, which are classified into three different categories based on risk or the potential to cause injury to patients using the devices.a The regulatory thresholds and controls for medical devices increase from class I to class III, with class III medical devices having the most rigorous regulatory controls.
- Class I devices have the lowest risk and demonstrate minimal potential to cause harm to a patient. These devices are the most basic devices and include bandages/medical patches, inhalers, pregnancy test kits, blood pressure monitors, and even blood glucose monitors, among others.
- Class II devices are considered advanced devices and present moderate risk to patients. These devices include in vitro diagnostic devices, such as blood serum immunological tests, next-generation sequencing tests, and blood hematology tests, as well as diagnostic imaging equipment, such as MRI and CT equipment, and surgical robots, to name a few.
- Class III constitutes high-risk devices that are used to sustain or support life. These devices are considered implantable devices and include orthopedic implants, cardiovascular implants (e.g., stents and pacemakers), contraceptive devices, and even breast implants.
After device conceptualization and during development of a prototype, understanding not only the different classes of medical devices but also which class your medical device falls into will aid in streamlining the regulatory process. For reference, you can access the FDA product classification database at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm which includes all registered medical devices, their classifications and product codes, and other regulatory information. If you are unaware of how to classify your medical device, you can also submit a 513(g) application to the FDA to get guidance regarding the classification of a device and whether the device is subject to 510(k) regulations.
----------------------------------
a The 21st Century Cures Act, enacted in 2016, further amended the FD&C Act to include provisions for how and when software is considered a medical device. Section 520(o) of the FD&C Act states that medical devices do not include software functions that are intended to support the administration of a healthcare facility; do not diagnose, cure, mitigate, prevent, or treat a disease or condition; transfer, store, convert formats, or display clinical laboratory tests or other device data/results, including electronic medical records; or acquire, process, or analyze medical images or signals from an in vitro diagnostic device or signal acquisition system.
Premarket Submission
After determination of the proper device classification, the medical device regulatory approval process begins with preparation of the appropriate premarket submission. However, before filing the appropriate premarket submission with the FDA, device manufacturers can submit a Pre-Submission (Pre-Sub). Pre-Subs, more commonly known now as Q-Submissions because each Pre-Sub is given a ‘Q’ identification number, are applications to the FDA that allow device manufacturers the opportunity to receive guidance from the FDA about the regulatory process and what the requirements are for a particular device. Even before the Pre-Sub, manufacturers can request an information meeting, during the FDA can provide feedback and questions that the manufacturers should consider when designing their devices, but the FDA is not obligated to answer any specific questions that the manufacturer has. The Pre-Sub should include a cover letter, description of the device, the information on the intended use of the device, any previous submissions if applicable, and a list of specific questions for the FDA. It is important to note that, while Pre-Subs are not required (i.e. device manufacturers can directly submit premarket submissions to the FDA without having a Pre-Sub, the Pre-Sub needs to be very specific in its intent). Another important note is that both Pre-Subs and informational meetings are different from 513(g) applications, as 513(g) applications are intended for inquiries about device classifications and what regulatory requirements are needed based on the classification. For more information about Pre-Sub guidance, refer to the FDA’s website at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/requests-feedback-and-meetings-medical-device-submissions-q-submission-program.
The most common premarket submissions are:
1. 510(k) – Premarket Notification
The 510(k) premarket notification is a Pre-Sub sent to the FDA that provides evidence not only of safety and effectiveness of the medical device but also that the device is substantially equivalent to a legally marketed device (commonly referred to as a predicate device). A medical device is considered substantially equivalent if the new device has the same intended use and the same technological characteristics as the predicate device; however, if the new device does not have the same technological characteristics as the predicate device, information (often times clinical data) demonstrating that the new device is as safe and effective as the predicate device must be submitted to the FDA via the 510(k) application. Legally marketed devices can either be pre-amendment devices, which are devices that were legally marketed in the U.S before the Medical Device Regulation Act (also known as the Medical Device Amendments) were enacted on May 28, 1976, or post-amendment devices, which are devices that were brought to the market through the premarket approval (PMA) or 501(k) pathway in accordance with the requirements of the Medical Device Amendments.
Before a medical device can be legally marketed in the U.S. under the 510(k) pathway, the party submitting the 510(k) application must receive a letter of substantial equivalence from the FDA. However, if the FDA does not deem the device to be substantially equivalent to a predicate device, a number of different mechanisms can be utilized:
- The applicant(s) can resubmit a 510(k) with new, pertinent data.
- The applicant(s) can request that the device be reclassified.
- The applicant(s) can submit a de novo classification request for class I or II designation.
- The applicant(s) can submit a PMA application.
Since substantial equivalence is an important part of the 510(k) process, the FDA has prepared a guidance document at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/510k-program-evaluating-substantial-equivalence-premarket-notifications-510k to update device developers and manufacturers, as well as FDA staff, about current review practices for 510(k) submissions.
Note that devices that proceed through the 510(k) pathway are “cleared” rather than “approved”. Therefore, once a 510(k) submission is cleared by the FDA, the device cannot be marketed as FDA-approved.
2. PMA – premarket approval
PMA is the process by which Class III medical devices are reviewed for their safety and effectiveness before the high-risk devices can be brought to market. PMA applications are significant more in-depth than a 510(k) application because the demonstration of safety and effectiveness of a class III device requires, typically, clinical studies and/or trials in humans to corroborate the data acquired from preclinical laboratory studies, all of which must be included in the application. The period from submission of a PMA application to approval is up to 180 days. Because clinical data are often required or strongly recommended to support a PMA, an investigational device exemption (IDE) application must be submitted to and approved by the FDA so that the device can be used in a clinical study on humans. A list of PMA guidance documents can be down on the FDA website at https://www.fda.gov/medical-devices/premarket-approval-pma/pma-guidance-documents.
3. De novo classification
The de novo classification provides an alternative pathway for classifying low-to-moderate risk (i.e. class I and II) devices. This enables device manufacturers or specification developers to seek FDA clearance without submitting a 510(k). De novo classification is appropriate for devices that either do not necessarily fit into a particular class of devices, are not equivalent to a legally marketed medical device, or have not been deemed substantially equivalent by the FDA following a 510(k) submission. For guidance on the acceptable review process for de novo classification request, visit the FDA website at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/acceptance-review-de-novo-classification-requests.
4. HDE – humanitarian device exemption
The HDE program was established by Congress under the Safe Medical Devices Act of 1990 as a means to facilitate the clearance or approval of devices for the treatment of orphan diseases, given that it is difficult to acquire enough clinical data on the use of the device for treating a rare disease that meets the standards of the FDA for evaluating safety and effectiveness under the traditional regulatory pathways. Applicants who are wishing to seek FDA regulatory approval for a humanitarian use device (HUD) can utilize the HDE pathway, so long as the HUD is intended for treatment or diagnosis of a disease with no more than 8000 individuals affected in the U.S. per year. The guidance document for the HDE program can be found here at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/humanitarian-device-exemption-hde-program.
Below is a breakdown of which premarket submissions are required for each device class per the FDA:
- Almost all Class I devices are exempt from premarket submission (pre-sub) and typically do not require obtaining FDA approval or clearance. However, some devices do require a 510(k) submission. All class I medical devices require compliance with current good manufacturing practices (cGMP) and need to meet the product registration requirements of class I devices.
- Most Class II devices require a 510(k) submission; however, 510(k) exemptions are available, depending on the particular medical device. You can find a list of class II exempt devices on the FDA’s website. In order for 510(k) submission to satisfy the FDA device registration requirements of class II devices, the applicant must demonstrate that the device is substantially equivalent to a legally marketed device.
- Most Class III devices require a PMA, the most stringent regulatory approval pathway.
After submitting the premarket submission, the FDA will review all applications, including a thorough examination of the data included in each application, and make a decision on whether or not to approve (or clear) the medical device. The FDA will then send a clearance letter (or PMA approval letter), confirming that the medical device can legally be marketed in the U.S.
FDA Regulation of Drugs
According to the FDA, a drug is defined as “intended for use in the diagnosis, cure, mitigation, treatment, or prevent of disease” and “an article—other than food—intended to affect the structure or any function of the body of man or other animals”. Compared to medical devices, achieving FDA regulatory approval of drugs is typically longer and more costly. On average, the time it takes for a drug to progress from the pre-clinical stage to the market can be on the order of 10–12 years, whereas the corresponding time for medical devices is on the order of 3–7 years. The total cost to bring medical devices to the market, including development, is estimated to be $30 million.b Comparably, the cost to develop and bring a new drug to market has increased exponentially over the past few decades, with some reports estimating that the total costs range from $1 to 3 billion. Furthermore, the majority of academic institutions are resource-constrained and do not have sufficient core competencies—i.e., funding, infrastructure, and personnel/expertise—needed to take a lead compound from the laboratory through clinical trials. This makes the commercialization of potential drugs incredibly difficult, even for small academic spinouts and startups, which is why drugs are frequently commercialized by medium-to-large pharmaceutical or biotechnology companies that have access to the resources needed to bring a drug through clinical trials and get marketed to the public. Below is an outline of the FDA drug approval process, beginning from preclinical development of an investigational new drug candidate to FDA approval and post-market surveillance (PMS).
Preclinical development
Upon identification of a lead drug candidate that has demonstrated therapeutic potential for a particular clinical indication, preclinical testing of that drug candidate is required. This stage of drug development entails evaluating the toxicity of the drug in non-human animals—often multiple species—to obtain basic information on the safety and efficacy of the drug candidate. In addition, during preclinical development, sponsors conduct formulation studies to determine the most effective means of administering the drug to the patient, including dosing and routes of administration. Pharmacology studies are also conducted to assess the ADME (absorption, distribution, metabolism, and excretion) properties of the drug in vivo in order to obtain a better understanding of the safety and efficacy of the drug candidate in humans. While this stage of drug development is not directly overseen by the FDA, the data acquired from preclinical investigations must be included in the Investigational New Drug application that is submitted to the FDA (see below).
Investigational New Drug (IND) Application
After reviewing the findings of preclinical studies of an investigational drug or biological product, the drug sponsor will decide whether the data are compelling enough to submit an IND application to the FDA, which is a requirement to conduct clinical studies on the drug in humans. However, INDs are not always needed for clinical investigations; the need for an IND depends on this specific intent of the investigation and the risk associated with the investigational drug. FDA guidance on whether an IND is required for clinical research studies in humans can be found here https://www.fda.gov/regulatory-information/search-fda-guidance-documents/investigational-new-drug-applications-inds-determining-whether-human-research-studies-can-be. Generally speaking, INDs are needed to initiate clinical investigations for: a new drug, a new method of use of an already-marketed drug (i.e. a new indication or new patient population), or a new drug in combination with another investigation drug or FDA-approved drug.
There are two categories of IND applications:
- Commercial: these applications are typically submitted by pharmaceutical and biotechnology companies seeking market approval of an investigational new drug.
- Research (non-commercial): these applications are submitted strictly for research studies by sponsors (typically an individual investigator, non-profit organization, or academic institution) and often result in publications in peer-reviewed journals. The majority of INDs filed are research INDs.
-------------
b The cost of development depends heavily on the class of the device and, therefore, whether the approval of the device requires acquisition of data from clinical trials/studies to demonstrate safety and effectiveness. This particular number is an estimate based on data to develop a class II, 510(k)-cleared medical device as of 2020.
There are three types of IND applications:
- Investigator IND: these applications are initiated and submitted by a phycisian who is responsible for conducting the clinical studies, often on behalf of a sponsor, such as a pharmaceutical company. Drug sponsors are responsible for selecting a qualified clinical investigator and for providing the investigator with the necessary information that they need to conduct the clinical investigation.
- Emergency Use IND: this type of IND is restricted to the authorization of an experimental drug for use in emergency situations involving life-threatening and/or severely debilitating diseases or conditions in which there is not sufficient time to prepare and submit an IND. Therefore, in these cases, the FDA can authorize the manufacturer to ship the investigational drug to investigators in advance of the IND submission.
- Treatment IND: these applications are for expanded use, which involves the use of an investigational new drug to treat specific patients or patient populations with serious/life-threatening conditions for which no comparative therapy is available. To qualify for a treatment IND, the investigational drug must meet the following four criteria:
- The sponsor of the drug is actively seeking FDA approval.
- The drug is intended for use in treating a life-threatening disease/condition.
- No satisfactory treatment is currently available for the intended indication.
- Clinical investigations are currently ongoing or have been completed.
What goes into an IND application?
- Animal pharmacology and toxicity data – this information is important to demonstrate that the investigational new drug is reasonably safe for testing in humans
- Chemistry, manufacturing, and control (CMC) information for the drug substance, drug product, placebo formulation, labelling, and environmental analysis to assess the impact of the investigational new drug on the environment
- Investigator information and clinical protocols – detailed information on how the clinical trials will be conducted; the qualifications of the investigator(s) listed on the IND application; and commitments of the sponsor and investigator(s) to obtain informed consent from participants of the trials, seek overview by an institutional review board (IRB), and abide by all government regulations for an investigational new drug
Importantly, the drug sponsor can seek FDA guidance at any stage of the IND process. Arguably, pre-IND is the important stage of the IND process when seeking FDA guidance. Seeking pre-IND guidance can be helpful and important to the drug sponsor in a variety of ways, including developing a strategy for drug development; obtaining guidance on the initiation of clinical trials; when seeking orphan designation, fast-track designation, accelerated approval, or approval of the drug under the animal efficacy rule; when there are no guidance documents available for the particular modality or entity; or simply requesting more information on the regulatory process for the drug candidate (assuming this information is not provided in existing guidance documents). However, development of an investigational drug product is the responsibility of the sponsor, and while the FDA can provide some guidance and oversight of the regulatory process, this guidance is limited due to resource constants at the agency. Similar to Q-Submissions, pre-IND meetings have a specific purpose. In the pre-IND meeting request, applicants should include: the meeting objective; agenda and proposed items for discussion; a list of specific, detailed questions; names of sponsor participants; composition of the drug; the proposed indication; dosing regimen; a proposed meeting date; and a timeline of when the pre-IND meeting packet will be submitted to the FDA (must be four weeks prior to the proposed meeting).
Consulting not only the FDA but also reputable external consultants with in-depth knowledge of the IND submission process and FDA regulatory standards on IND submissions is crucial because IND applications are highly technical documents, and a lack of clarity, organization, data, and explanation of the preclinical results can often result in a clinical hold or even a technical rejection of the application. A complete IND application can be hundreds of pages long and take several months to compile, depending on the data required to demonstrate that the drug is safe enough to administer to humans and effective for the intended indication. Therefore, it is important for the sponsor or whomever is submitting the IND to be mindful of the requirements (and pitfalls) when preparing the application.
Upon submission of the complete IND application, the FDA has 30 days to review the application, during which the clinical investigators are not allowed to conduct clinical studies in humans. After the 30-day period, the IND goes into effect, unless the FDA puts a partial or complete hold on the IND following review.
At this point, the drug manufacturer is permitted to ship the investigational new drug to the investigator(s) named in the IND.
A list of guidance documents for preparing the IND application can be found on the FDA’s website at https://www.fda.gov/drugs/types-applications/investigational-new-drug-ind-application#FDA%20Guidances%20for%20Investigational%20New%20Drugs.
Clinical Trials
Once the FDA has given authorization to an IND sponsor to conduct clinical investigations, the research studies outlined in the IND, commonly known as clinical trials, can begin. These trials are conducted in three phases: Phases I, II, and III, with the first being Phase I. The investigator (or sponsor-investigator, if the sponsor and investigator are the same person) is responsible for conducting the clinical trials—this is the person or team of people who administers the drug.
- Phase I emphasizes safety. In Phase I, approximately 20–80 patients participate. During this phase, investigators monitor the side effects of the drug and study how the drug is metabolized and excreted from the body. Dosage tolerance is also assessed to determine the maximum tolerated dose of the drug without producing deleterious side effects. Of all three phases, Phase I has the highest success rate, with approximately 70% of drugs moving to Phase II.
- Phase II emphasizes the effectiveness of the drug for the intended indication. In Phase II, hundreds of patients with the particular disease or condition participate. The ultimate goal of Phase II is to determine if the drug ultimately improves the disease. During this phase, the dosing regimen is studied to determine the most effective frequency of administration, the dose per single administration, the route of administration, and even the treatment durations. The majority of Phase II trials are randomized, which means patients can either be given the experimental drug(s) or a placebo (control). The randomizations are commonly double-blind, which means neither the investigator nor the patient knows what they are receiving. Approximately 1/3 of drugs that enter this phase pass and move on to Phase III. After completion of Phase II, the FDA and the drug sponsor will assess the data and discuss whether and how Phase III studies will be conducted.
- Phase III is intended to gather substantially more data on both safety and effectiveness of the drug in a larger population size (1000’s of participants). These studies are typically both randomized and blinded (either single or double), and the majority of the safety information that will go into the packaging and labelling of the drug will be generated during Phase III. Since Phase III clinical studies can last for 1–4 years, long-term effects can often be observed during this phase. Approximately 25% of drugs that enter Phase III will move on to seek FDA approval.
New Drug Application
After completion of Phase III clinical trials, the FDA meets with the drug sponsor to discuss the results of the clinical trials prior to submission of a New Drug Application (NDA), which is a formal application to request permission from the FDA to manufacture and market a drug in the US. This pre-NDA meeting should occur no less than 60 days before submission of an NDA. Furthermore, 30 days before the pre-NDA meeting, a briefing package, which contains information about the proposed indication, a summary of the safety and effectiveness data acquired during clinical trials, CMC information, the proposed format of the NDA submission, and a timeline of the application submission, must be submitted to the FDA.
An NDA is submitted to the Center for Drug Evaluation and Research (CDER), a division of the FDA that regulates prescription, over-the-counter, and generic drugs, including some biologics, such as monoclonal antibodies, immunomodulators, and even therapeutic proteins. However, all other biologics are regulated by a different division of the FDA known as the Center for Biologics Evaluation and Research (CBER). The NDA is a comprehensive document that tells the full story of the drug’s development and includes all data from preclinical testing in animals and human Phase I–III clinical trials; information about how the drug behaves in the human body; labelling information; information on the manufacturing and processing of the drug, including the facilities where the manufacturing will take place; the indication for which the drug will be used; and all of the chemical information about the drug. An NDA can often be 100,000+ pages in length, and the prescription drug user fee for filing an NDA in 2024 is approximately $4 million, if the application requires clinical data.
Upon receipt of the NDA, the FDA is required to conduct a preliminary review of the application within 60 days. If the FDA determines that the NDA is sufficiently comprehensive, it will file the NDA for either standard review (10 months) or priority review (6 months). During the review, experts, often as part of an advisory panel, will review all of the clinical data, the proposed labelling of the drug, and even inspect the facilities where the drug will be manufactured. Upon approval of the NDA, the sponsor of the drug is free to manufacture and market the drug in the US. If the FDA denies approval, it will send a complete response letter to the sponsor that entails the specific deficiencies that led to the denial and recommendations for resubmission.
If the (investigational new) drug does not qualify for an NDA, two other applications are available to seek regulatory approval:
- Biologic License Application (BLA) – biologics, such as vaccines and recombinant proteins, that are used in medical treatments are approved by the FDA through a BLA and are evaluated by the CBER.
- Abbreviated New Drug Application (ANDA) – generic drugs previously approved for marketing via an NDA submitted by another manufacturer are approved through an ANDA, which does not require the completion of clinical trials.
Post-Market Surveillance (Phase IV)
Approval of a new drug to be legally marketed in the US means that scientific and regulatory experts, after reviewing thousands of pages of clinical data, have determined that there is substantial evidence that a drug is safe and provides a meaningful therapeutic benefit in treating a particular clinical indication. However, Phase I–III clinical trials can only assess the safety in a few thousand participants. Oftentimes, the FDA will require drug manufacturers to conduct a Phase IV trial, commonly known as post-market surveillance (PMS), as a condition of FDA approval. Phase IV trials serve to evaluate the real-word effectiveness of a drug once it becomes available to the “public”, as the exposure of the drug to hundreds of thousands of patients of different ethnicities, ages, and other identities will yield the true safety profile of the drug and provide a more comprehensive indication of its effectiveness.